This tutorial shows how to start the FastRet GUI and use its four modes.
Starting the GUI
Install the package and run this in an interactive R session:
FastRet::start_gui()The console prints:
Listening on http://localhost:8080Open http://localhost:8080 in your browser to use the GUI.
The GUI has four modes, shown as tabs at the top: Train, Select, Adjust and Predict. Hover over a tab to see its full name. Click the question-mark icon next to any input for a short help text.
If start_gui() reports a CDK version error, FastRet
needs CDK 2.9 or newer and a working Java installation. See the Installation
article for how to set up Java and rcdk.
Train new Model
Use the Train tab to build a model from your own
measurements: an Excel file with the names, SMILES and retention times
of metabolites measured on your column. FastRet ships an example file
with 458 metabolites measured on a reverse-phase column (35 °C, 0.3
ml/min). Download
RP.xlsx to upload it directly in the (web) GUI — no R
installation required. If you have the package installed, the same file
is available via system.file():
path <- system.file("extdata", "RP.xlsx", package = "FastRet")
cat(path, "\n", sep = "")
#> /home/runner/work/_temp/Library/FastRet/extdata/RP.xlsx
df <- openxlsx::read.xlsx(path, 1)
head(df)
#> RT SMILES NAME INCHIKEY
#> 1 0.90 C(CN)CN 1,3-DIAMINOPROPANE XFNJVJPLKCPIBV-UHFFFAOYSA-N
#> 2 0.90 C(CCNCCCN)CNCCCN SPERMINE PFNFFQXMRSDOHW-UHFFFAOYSA-N
#> 3 0.91 C(CCNCCCN)CN SPERMIDINE ATHGHQPFGPMSJY-UHFFFAOYSA-N
#> 4 0.91 C(CN)CNCCCN BIS(3-AMINOPROPYL)AMINE OTBHHUPVCYLGQO-UHFFFAOYSA-N
#> 5 0.93 CC(=O)NCCCNCCCCNCCCN N1-ACETYLSPERMINE GUNURVWAJRRUAV-UHFFFAOYSA-N
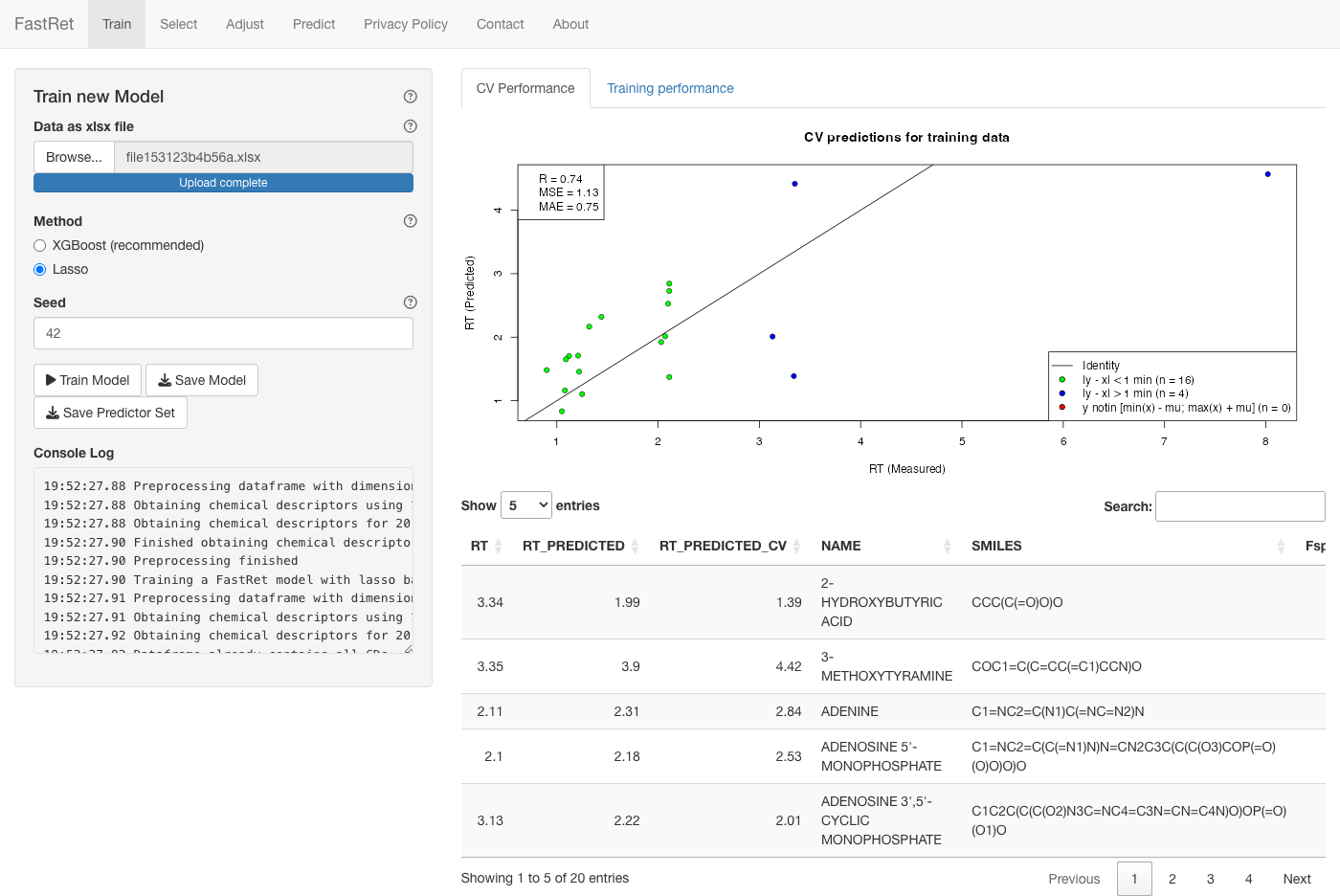
#> 6 0.98 C1=C(NC=N1)CCN HISTAMINE NTYJJOPFIAHURM-UHFFFAOYSA-NSet the controls, then click Train Model:
-
Data as xlsx file — your Excel file (columns
RT,NAME,SMILES). - Method — XGBoost (recommended) or Lasso.
-
Seed — random seed. Keep the default
(
42) for reproducible results.
The Console Log shows progress. When training finishes, the main panel shows the cross-validation and training plots and a table of predictions. For the details, see Model-Training in the Package-Internals article.
Two buttons then appear: Save Model (an
.rds file) and Save Predictor Set (the
training data with descriptors). To predict with your model, save it,
switch to the Predict tab, upload it there, enter your
SMILES and click Predict (see Predict Retention Times).
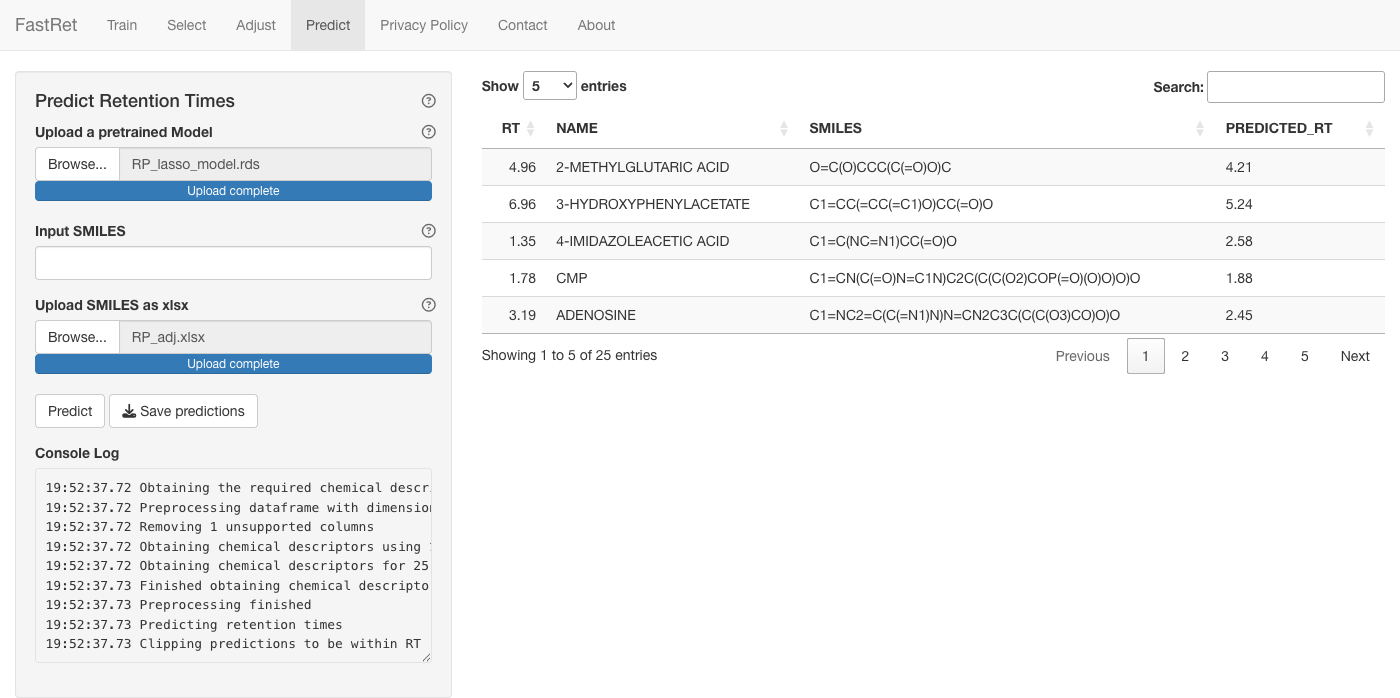
Predict Retention Times
Use a saved model to predict retention times for new molecules. To
try it without training first, download the example model RP_lasso_model.rds
(a LASSO model trained on the example RP.xlsx).
- Upload the model under Upload a pretrained Model.
- Enter a SMILES in Input SMILES, or upload an Excel file
(columns
NAME,SMILES) under Upload SMILES as xlsx. - Click Predict.
The predictions appear as a table in the main panel. Click Save predictions to download them as an Excel file.
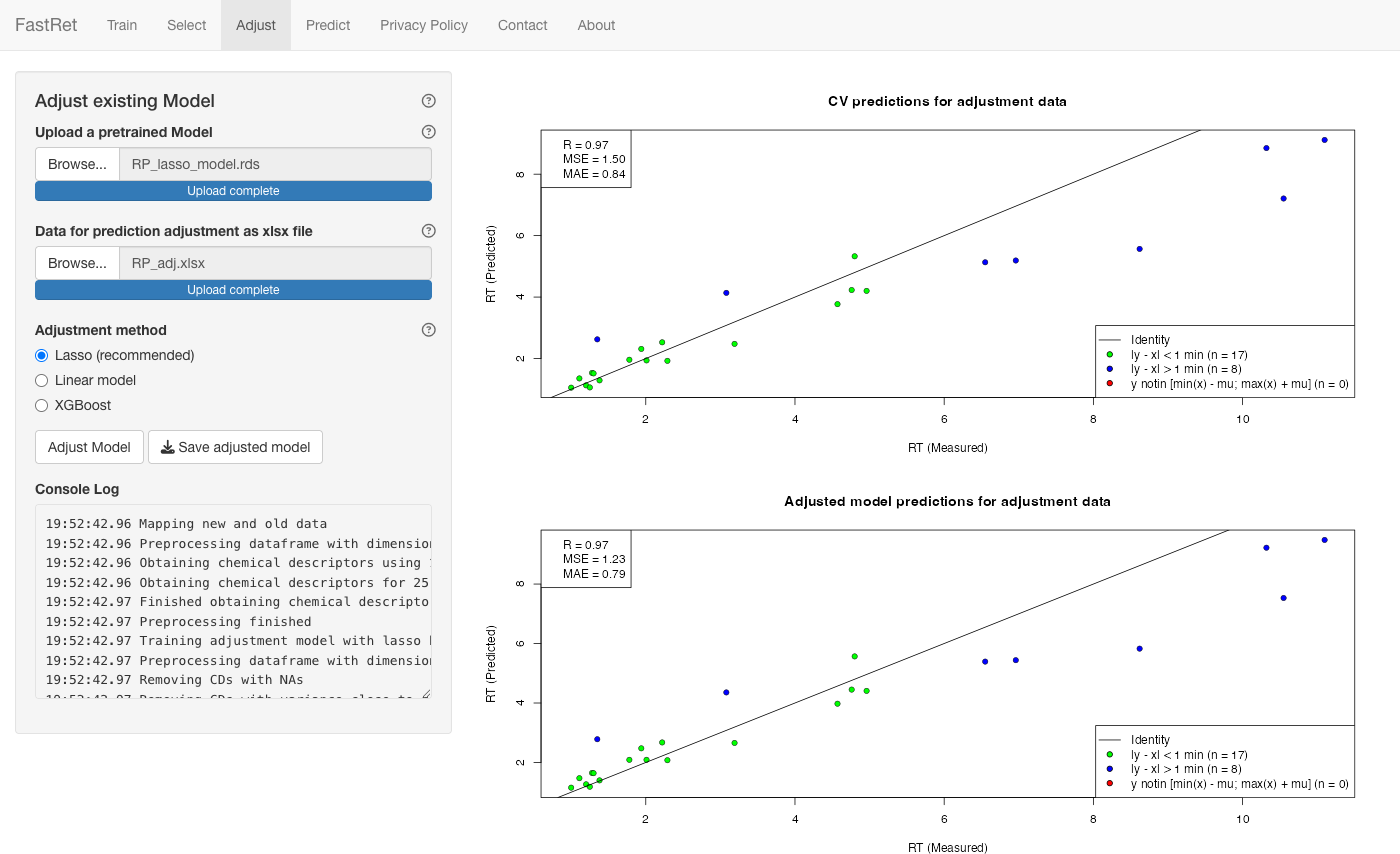
Adjusting existing model
If you re-measured some metabolites on a new column that were also
measured on the original one, use the Adjust tab to
adapt an existing model to the new column. An example re-measurement
file (25 metabolites from RP.xlsx measured under a steeper
gradient) is available as RP_adj.xlsx.
- Upload the model under Upload a pretrained Model.
- Upload the re-measured metabolites (columns
RT,NAME,SMILES) under Data for prediction adjustment as xlsx file. - Choose the Adjustment method: Lasso (recommended), Linear model or XGBoost. Lasso and XGBoost use the base retention time and the molecular descriptors, so they can capture compound-specific shifts; the linear model fits a straight-line correction from the base retention time alone.
- Click Adjust Model.
The main panel shows the performance of the adjusted model. Click Save adjusted model to download it. Use it like any other model on the Predict tab.
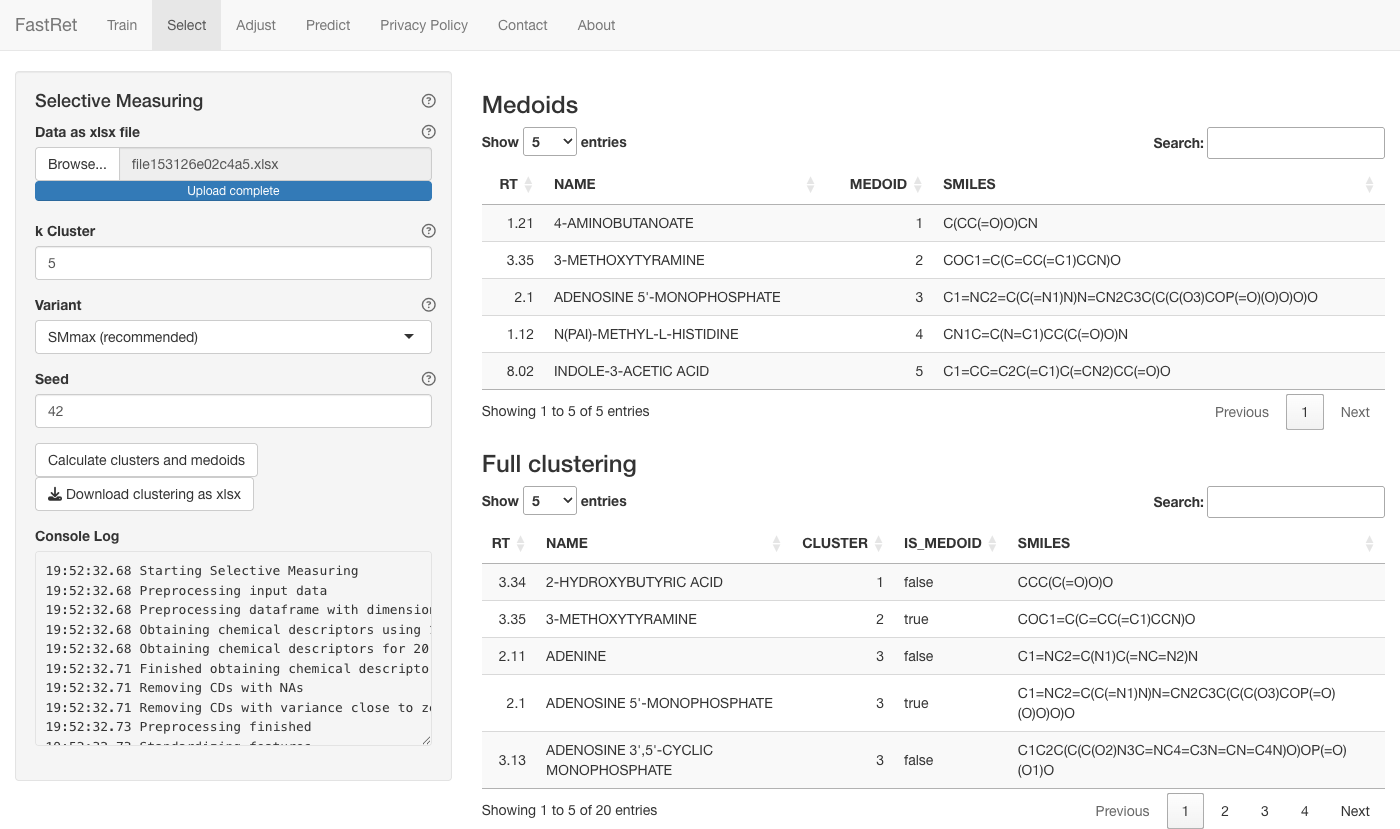
Selective Measuring
Adjusting a model to a new column needs a few metabolites measured on
that column. The Select tab picks the k
most representative molecules to measure, so you cover the diversity of
your dataset with as little lab work as possible. It uses Ridge
Regression and PAM (k-medoids) clustering.
- Upload an Excel file (columns
NAME,SMILES,RT). - k Cluster — how many molecules to select.
- Variant — how strongly the retention time guides the selection: SMmax (default, weighted like the most important descriptor), SM1 (unscaled), SM0 (excluded) or SMinf (retention time only). The info button explains each.
-
Seed — random seed. Keep the default
(
42) for a reproducible selection. - Click Calculate clusters and medoids.
- Click Download clustering as xlsx to save the selected molecules and their clusters.
The main panel lists the selected molecules and the full clustering. Measure the selected molecules on the new column, then use them to adjust your model.