This article shows how Metabodecon can be used for deconvoluting and aligning one-dimensional NMR spectra using the pre-installed Sim dataset as an example. The Sim dataset includes 16 simulated spectra, each with 2048 data points ranging from ≈ 3.6 to 3.3 ppm. These simulated spectra closely mimic the resolution and signal strength of real NMR experiments on blood plasma from 16 individuals. The Sim dataset is used instead of the Blood dataset because it is smaller, faster to process, and comes pre-installed with the package. For more information on the Sim and Blood datasets, see Datasets.
Deconvolute spectra
To find the path to the Sim dataset, you can use the
metabodecon_file() function, which returns the path to any
file or directory within the package directory. To deconvolute the
spectra within the Sim dataset you can read them into R using
read_spectra() and then call deconvolute() as
follows:
sim_dir <- metabodecon::metabodecon_file("bruker/sim")
sim <- metabodecon::read_spectra(sim_dir)
deconvoluted_spectra <- metabodecon::deconvolute(
sim, # The object containing spectra
sfr = c(3.35, 3.55), # Borders of signal free region (SFR) in ppm
smopts = c(2, 5), # Smoothing parameters
verbose = FALSE, # Disable verbose output
ask = TRUE # Enable interactive prompting
)After calling deconvolute(), the function will ask you
to answer the following questions to determine the optimal deconvolution
parameters interactively:
- Use same parameters for all spectra? (y/n)
- Number of spectrum for adjusting parameters? (1: sim_01, 2: sim_02, …)
- Signal free region correctly selected? (y/n)
- Water artefact fully inside red vertical lines? (y/n)
You can answer questions one and two with y and
1, as the dataset is homogeneous, i.e., all spectra were
measured in the same lab with the same acquisition and processing
parameters. However, for heterogeneous datasets, it’s advisable to
optimize parameters for each batch of spectra individually.
Questions three and four are accompanied by two plots, shown in Figure 1, which help you to verify the
accuracy of the selected signal-free region (SFR) and water signal
half-width (WSHW) 1. In this case, the provided parameters are
already fine, so you can answer both questions with y. If
adjustments are needed, you can respond with n and input
the correct values.

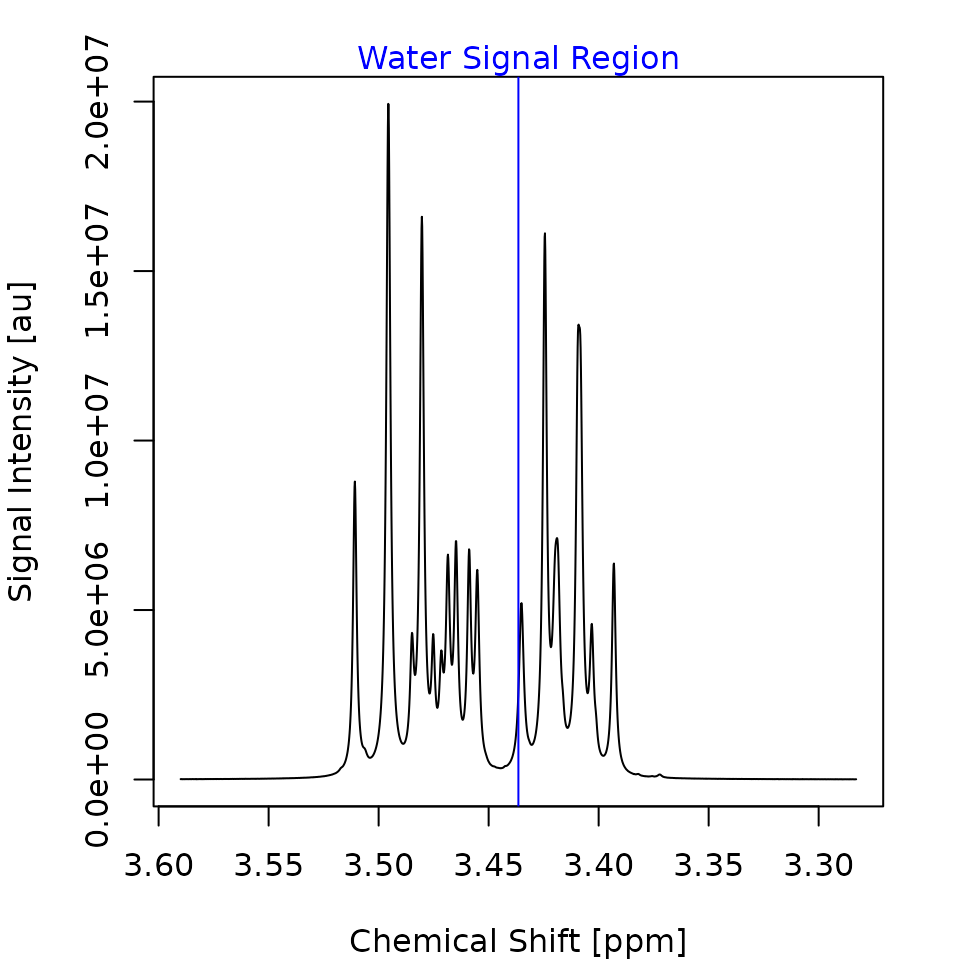
Figure 1. The first spectrum of the Sim dataset. The x-Axis gives the chemical shift of each datapoint in parts per million (ppm). The y-Axis gives the signal intensity of each datapoint in arbitrary units (au). The signal free regions are shown as green rectangles in the left plot. The water signal region, usually shown as blue rectangle, is shown in the right plot. Because the water signal half width is set to zero in this case, the rectangle collapses to a vertical line.
When using the function in scripts, where interactive user input is
not desired, you can disable the interactive prompting by setting
parameter ask to FALSE. In this case, the
provided parameters will be used for the deconvolution of all spectra
automatically. 2
Visualize deconvoluted spectra
After completing the deconvolution, it is advisable to visualize the
extracted signals using plot_spectrum() to assess the
quality of the deconvolution.
# Visualize the first spectrum.
metabodecon::plot_spectrum(deconvoluted_spectra[[1]])
# Visualize the second spectrum, this time without the legend.
metabodecon::plot_spectrum(deconvoluted_spectra[[1]], lgd = FALSE)
# Visualize all spectra and save them to a pdf file
pdfpath <- tempfile(fileext = ".pdf")
pdf(pdfpath)
for (x in deconvoluted_spectra) {
metabodecon::plot_spectrum(x, main = x$filename)
}
dev.off()
cat("Plots saved to", pdfpath, "\n")Out of the 16 generated plots, the first two are shown as examples in Figure 2. Things to look out for are:
- That the smoothing does not remove any real signals. If the
smoothing is too strong, i.e., the smoothed signal intensity (SI) is
very different from the raw SI, you should adjust the smoothing
parameters
smoptsin the call togenerate_lorentz_curves(). - That the superposition of the lorentz curves is a good approximation
of the smoothed SI. If major peaks are missed by the algorithm, you
should reduce the threshold
deltain the call togenerate_lorentz_curves().
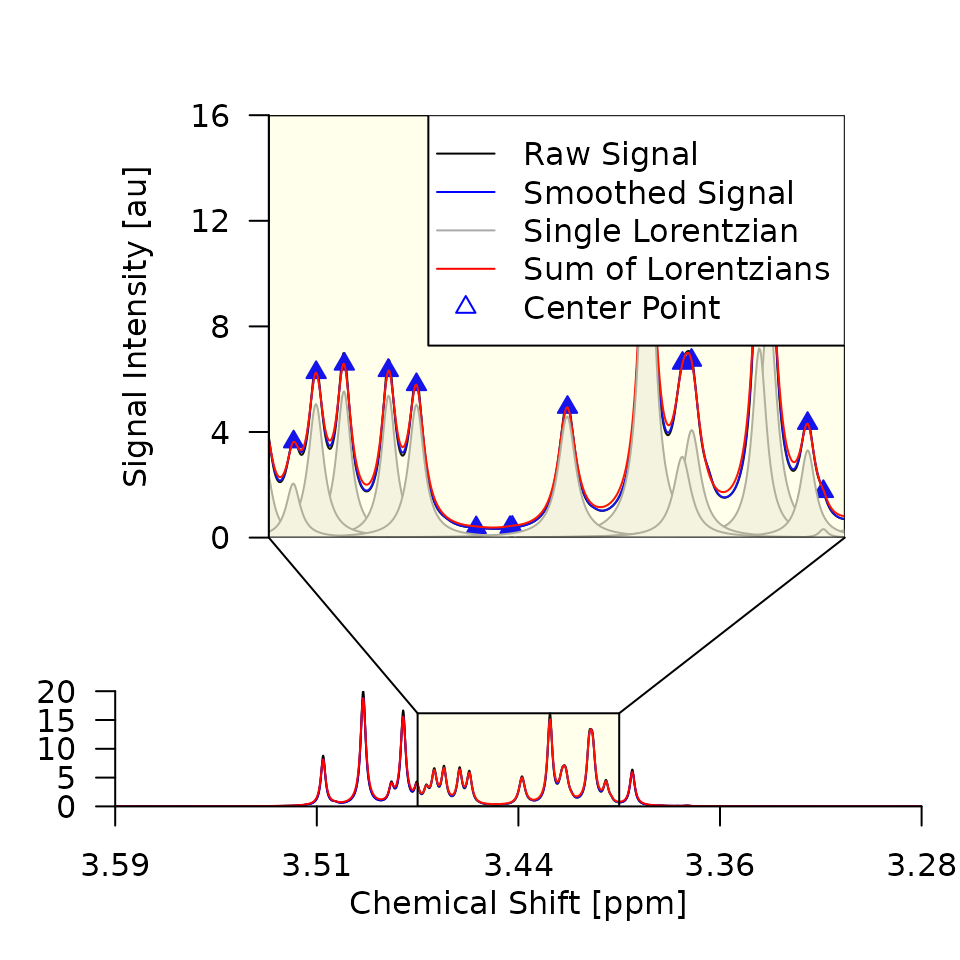
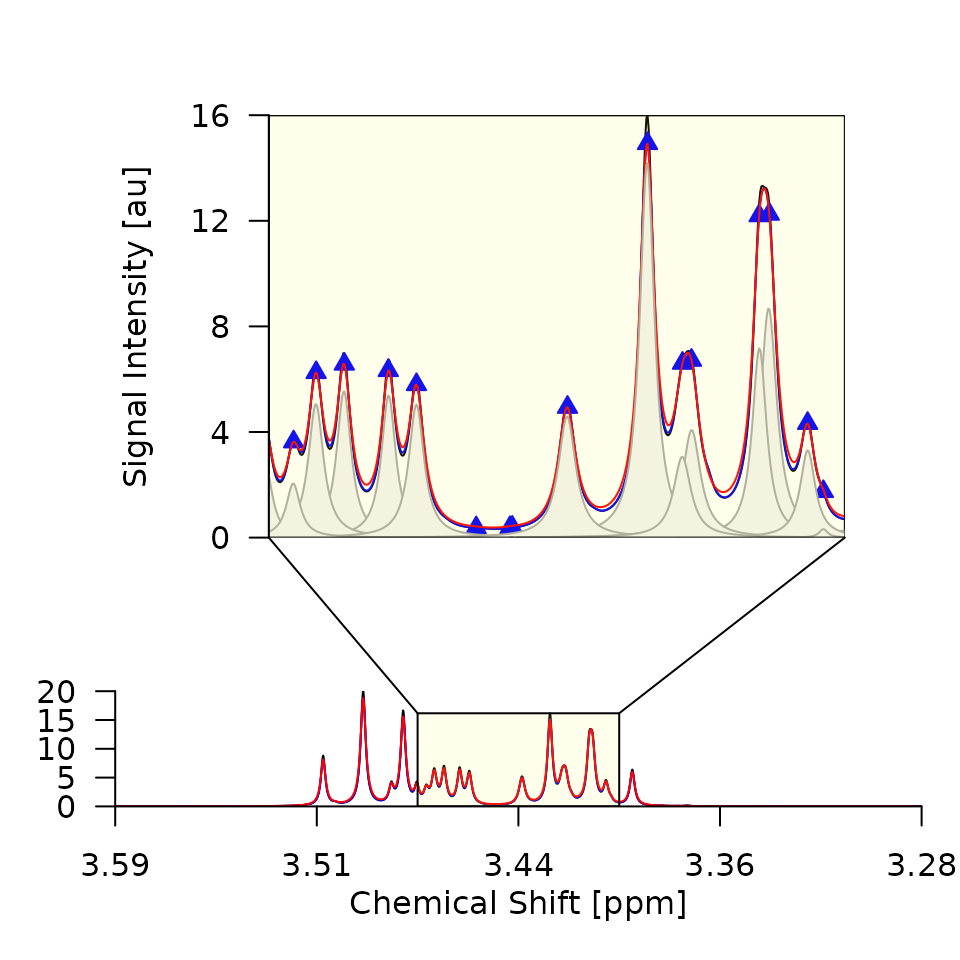
Figure 2. Deconvolution results for the first two
spectra of the Sim dataset. The raw SI (black), smoothed SI (blue), and
superposition of Lorentz curves (red) are closely aligned, indicating
that smopts and delta were chosen well and
that the deconvolution was successful.
Align deconvoluted spectra
The last step in the Metabodecon Workflow is to align the deconvoluted spectra. This is necessary because the chemical shifts of the peaks in the spectra may vary slightly due to differences in the measurement conditions.
To perform the alignment, you can use align(). To
visualize the data before and after the alignment, you can use
plot_spectra():
# Plot spectra before alignment. Only show spectra 1-8 for clarity.
metabodecon::plot_spectra(deconvoluted_spectra[1:8], lgd = FALSE)
# Align spectra and plot again.
aligned_spectra <- try(metabodecon::align(deconvoluted_spectra)) # (1)
metabodecon::plot_spectra(aligned_spectra[1:8])
# (1) The call to align() is wrapped in try() because the function may fail
# if speaq's Bioconductor dependencies (MassSpecWavelet, impute) are missing
# and the code runs in a non-interactive R session (e.g., during vignette
# creation). In interactive sessions, try() is not needed, as the user will
# be prompted to install missing dependencies automatically.The resulting plots are shown in Figure 3. Before the alignment, the spectra exhibit generally similar shapes but do not perfectly overlap. After the alignment, the spectra are much more consistent with each other, indicating that the alignment was successful. Notably, spectrum two has been shifted significantly to the left.
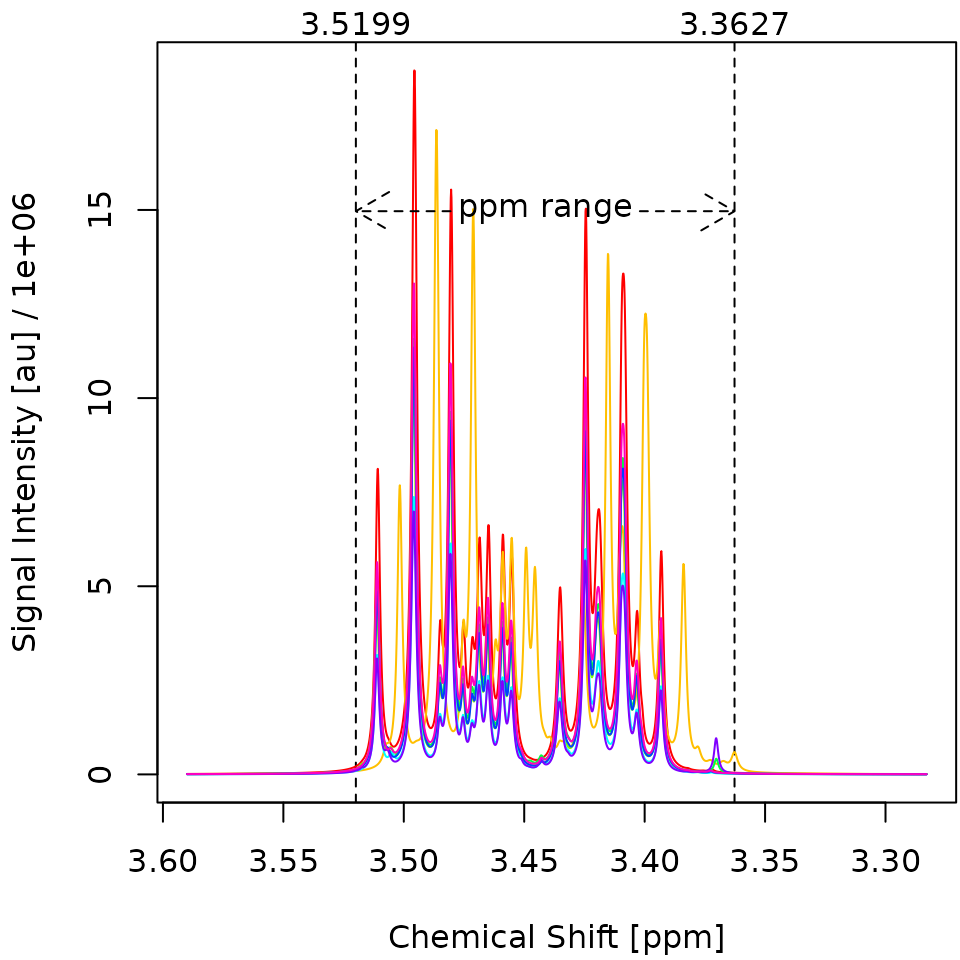
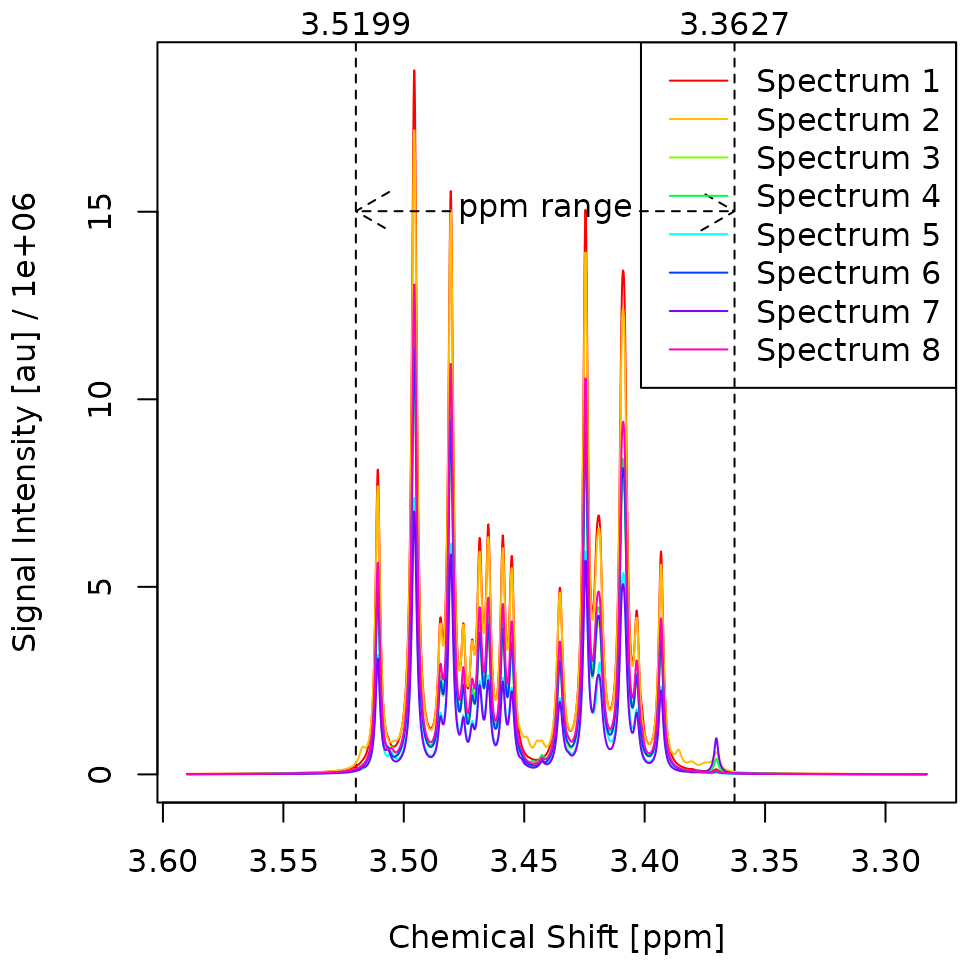
Figure 3. Overlay of the first eight deconvoluted spectra from the Sim dataset before alignment (left) and after alignment (right). The x-Axis gives the chemical shift of each datapoint in parts per million (ppm). The y-Axis gives the signal intensity of each datapoint in arbitrary units (au). All specta are pretty similar to each other except for Spectrum 2, which got shifted approx. 0.01 ppm to the right.