Automatic deconvolution of a 1D NMR spectrum into several Lorentz curves and the integration of them. The NMR file needs to be in Bruker format or jcamp-dx format.
This function has been deprecated with metabodecon version v1.2.0 and will be
removed with version 2.0.0. Please use deconvolute() instead.
![[Deprecated]](figures/lifecycle-deprecated.svg)
Arguments
- filepath
Complete path of the file folder (Notice for Bruker format: filepath needs to be the spectrum folder containing one or more different spectra (e.g."C:/Users/Username/Desktop/spectra_from_bruker"))
- filename
Name of the NMR file. (Notice for Bruker format: filename need to be the name of your spectrum which is also the name of the folder) (Default: filename = NA to analyze more spectra at once)
- file_format
Format (bruker or jcampdx) of the NMR file. (Default: file_format = "bruker")
- number_iterations
Number of iterations for the approximation of the parameters for the Lorentz curves (Default: number_iterations=10)
- range_water_signal_ppm
Half width of the water artefact in ppm (Default: range_water_signal=0.1527692 (e.g. for urine NMR spectra))
- signal_free_region
Row vector with two entries consisting of the ppm positions for the left and right border of the signal free region of the spectrum. (Default: signal_free_region=c(11.44494, -1.8828))
- smoothing_param
Row vector with two entries consisting of the number of smoothing repeats for the whole spectrum and the number of data points (uneven) for the mean calculation (Default: smoothing_param=c(2,5))
- delta
Defines the threshold value to distinguish between signal and noise (Default: delta=6.4)
- scale_factor
Row vector with two entries consisting of the factor to scale the x-axis and the factor to scale the y-axis (Default: scale_factor=c(1000,1000000))
- debug
Logical value to activate the debug mode (Default: debug=FALSE)
- store_results
Specifies whether the lorentz curve parameters
A,lambdaandx_0and the approximated spectrum should be stored on disk (in addition to returning them). Ifstore_resultsisNULL(default), the user is asked interactively where the files should be stored. If FALSE, the results are not stored. If TRUE, the results are stored in a subdirectory of R's per-session temporary directory.
Value
A decon0 object as described in Metabodecon Classes.
References
Haeckl, M.; Tauber, P.; Schweda, F.; Zacharias, H.U.; Altenbuchinger, M.; Oefner, P.J.; Gronwald, W. An R-Package for the Deconvolution and Integration of 1D NMR Data: MetaboDecon1D. Metabolites 2021, 11, 452. https://doi.org/10.3390/metabo11070452
Author
2020-2021 Martina Haeckl: initial version.
2024-2025 Tobias Schmidt: Minor updates to pass CRAN checks. Parameters
debug and store_results added.
Examples
## ATTENTION: using MetaboDecon1D() for deconvolution is deprecated. Please use
## deconvolute() instead.
## The following example shows how a subset of the Sim dataset, consisting
## of two spectrum objects, can be deconvoluted using `MetaboDecon1D()`. The
## whole example code is wrapped into `evalwith()` to simulate user input.
## When using the function interactively, you should type in the answers to
## the questions manually.
expected_answers <- c(
"10", # Subfolder of your filepath, i.e. the experiment number?
"10", # Subsubsubfolder of filepath, i.e. the processing number?
"y", # Use same parameters for all spectra?
"1", # File to adjust all parameters.
"n", # Signal free region borders correct selected?
"3.55", # Left border.
"3.35", # Right border.
"y", # Signal free region borders correct selected?
"n", # Water artefact fully inside red vertical lines?
"0", # Half width range (in ppm) for the water artefact.
"y", # Water artefact fully inside red vertical lines?
"n" # Save results as text documents?
)
sim <- metabodecon_file("bruker/sim_subset")
evalwith(answers = expected_answers, {
sim_decon <- MetaboDecon1D(sim)
})
#> What is the name of the subfolder of your filepath:
#> [e.g. 10 for C:/Users/Username/Desktop/spectra_folder/spectrum_name/10]
#> 10
#> What is the name of the subsubsubfolder of your filepath:
#> [e.g. 10 for C:/Users/Username/Desktop/spectra_folder/spectrum_name/10/pdata/10]
#> 10
#> Do you want to use the same parameters (signal_free_region, range_water_signal_ppm) for all spectra? (y/n)
#> y
#> [1] "sim_01" "sim_02"
#> Choose number of file which is used to adjust all parameters: [e.g. 1]
#> 1
#> The selected file to adjust all parameters for all spectra is: sim_01
#> Start deconvolution of sim_01:
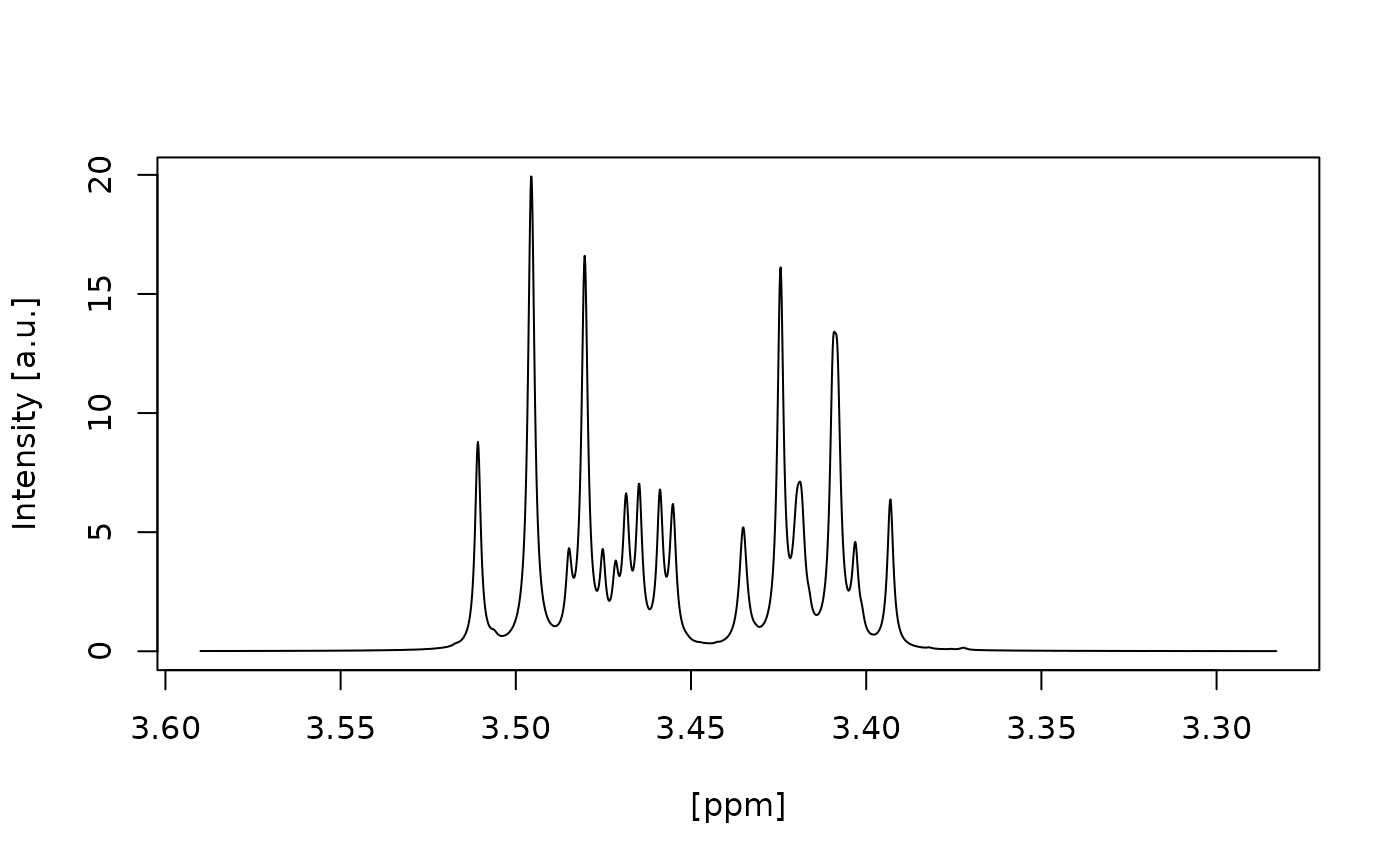 #> Signal free region borders correct selected? (Area left and right of the green lines) (y/n):
#> n
#> Choose another left border: [e.g. 12]
#> 3.55
#> Choose another right border: [e.g. -2]
#> 3.35
#> Signal free region borders correct selected? (Area left and right of the green lines) (y/n):
#> n
#> Choose another left border: [e.g. 12]
#> 3.55
#> Choose another right border: [e.g. -2]
#> 3.35
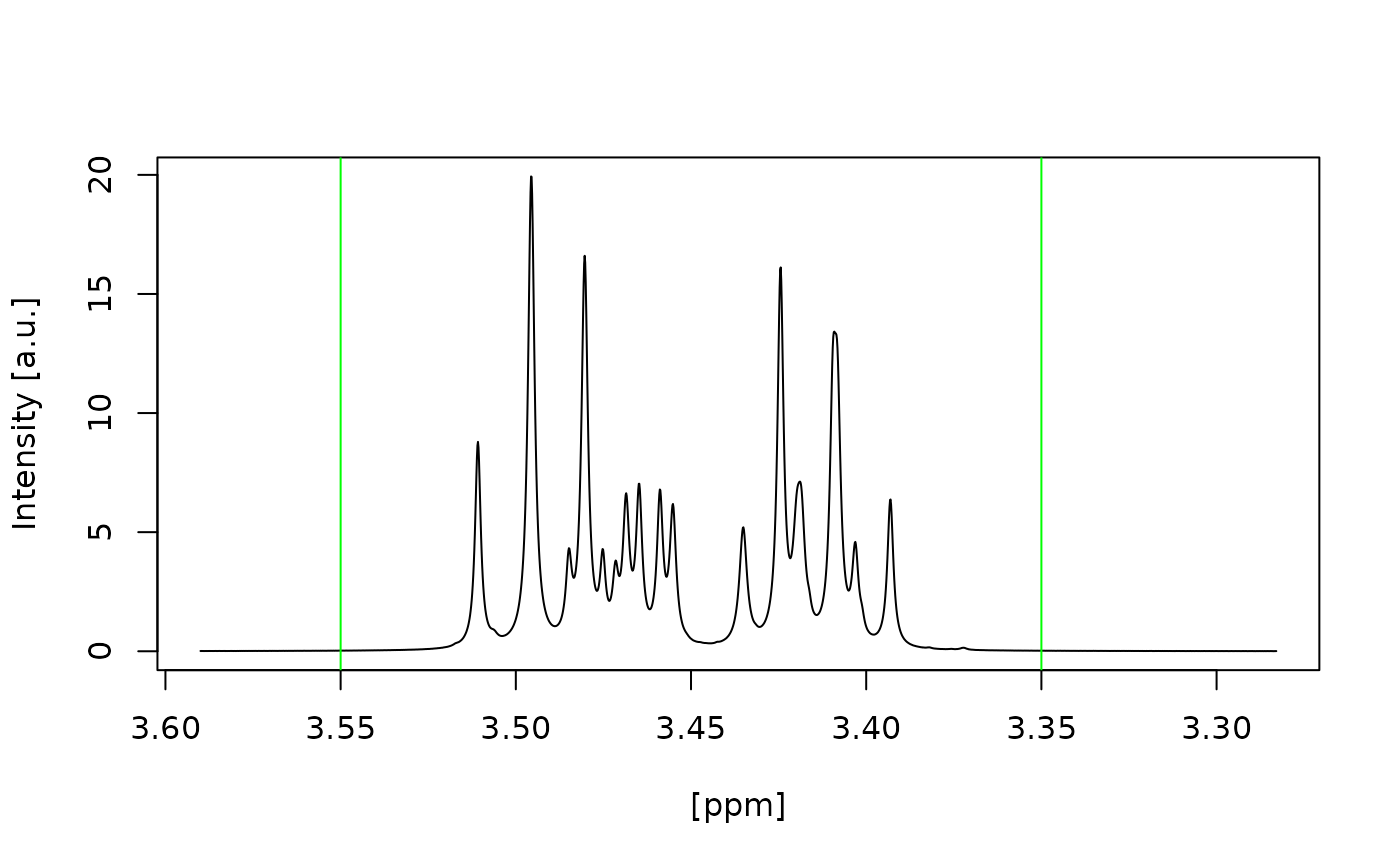 #> Signal free region borders correct selected? (Area left and right of the green lines) (y/n):
#> y
#> Signal free region borders correct selected? (Area left and right of the green lines) (y/n):
#> y
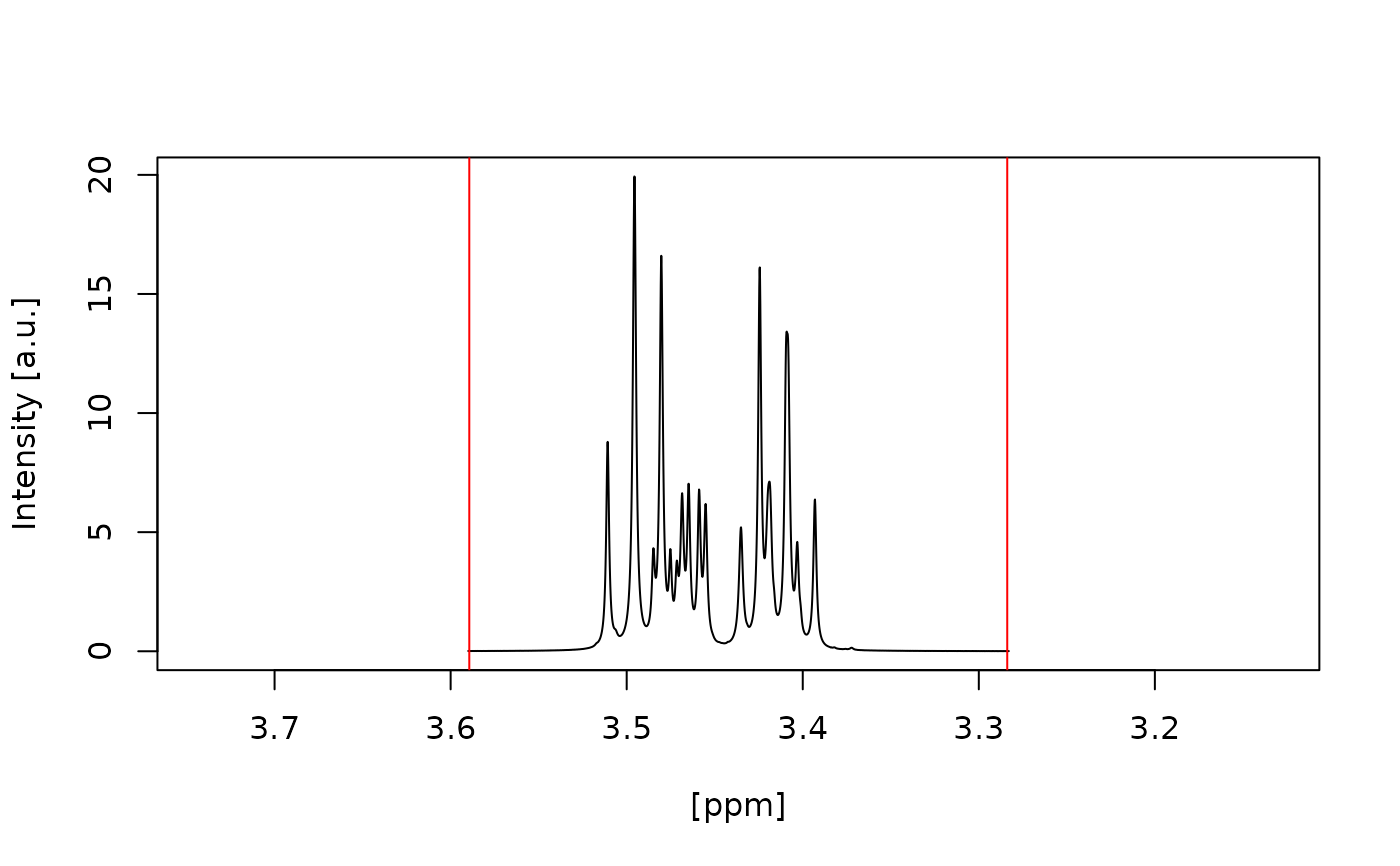 #> Water artefact fully inside red vertical lines? (y/n):
#> n
#> Choose another half width range (in ppm) for the water artefact: [e.g. 0.1222154]
#> 0
#> Water artefact fully inside red vertical lines? (y/n):
#> n
#> Choose another half width range (in ppm) for the water artefact: [e.g. 0.1222154]
#> 0
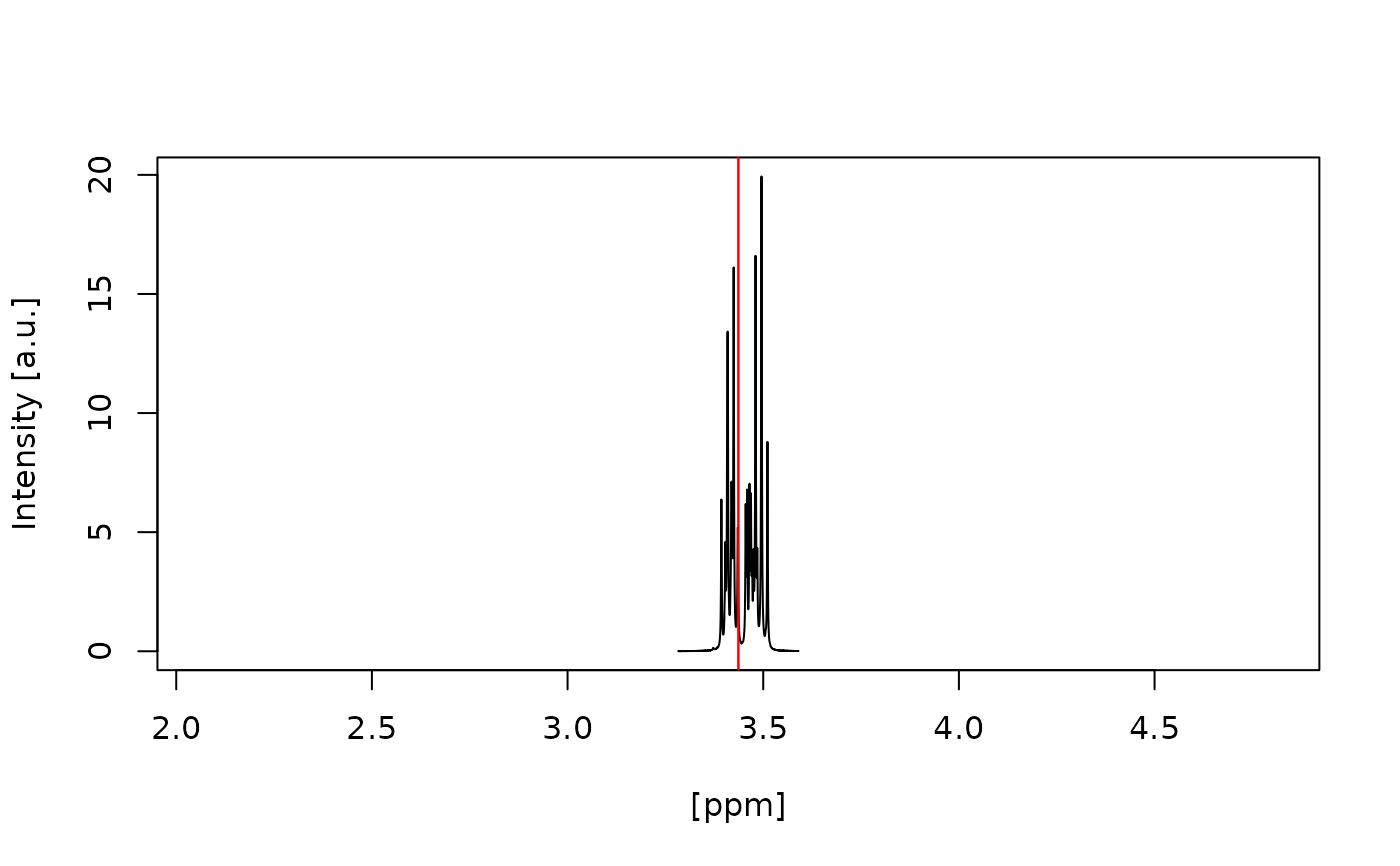 #> Water artefact fully inside red vertical lines? (y/n):
#> y
#>
#> Normed MSE value of iteration 1 is:
#> [1] 4.996559e-09
#>
#> Normed MSE value of iteration 2 is:
#> [1] 2.17297e-09
#>
#> Normed MSE value of iteration 3 is:
#> [1] 1.778563e-09
#>
#> Normed MSE value of iteration 4 is:
#> [1] 1.652956e-09
#>
#> Normed MSE value of iteration 5 is:
#> [1] 1.571827e-09
#>
#> Normed MSE value of iteration 6 is:
#> [1] 1.539285e-09
#>
#> Normed MSE value of iteration 7 is:
#> [1] 1.507361e-09
#>
#> Normed MSE value of iteration 8 is:
#> [1] 1.491203e-09
#>
#> Normed MSE value of iteration 9 is:
#> [1] 1.476108e-09
#>
#> Normed MSE value of iteration 10 is:
#> [1] 1.460549e-09
#> Save results as text documents at /home/runner/work/_temp/Library/metabodecon/example_datasets/bruker/sim_subset? (y/n)
#> n
#> Skipping saving of results.
#> Start deconvolution of sim_02:
#>
#> Normed MSE value of iteration 1 is:
#> [1] 4.449633e-09
#>
#> Normed MSE value of iteration 2 is:
#> [1] 1.688869e-09
#>
#> Normed MSE value of iteration 3 is:
#> [1] 1.276977e-09
#>
#> Normed MSE value of iteration 4 is:
#> [1] 1.135132e-09
#>
#> Normed MSE value of iteration 5 is:
#> [1] 1.085964e-09
#>
#> Normed MSE value of iteration 6 is:
#> [1] 1.056171e-09
#>
#> Normed MSE value of iteration 7 is:
#> [1] 1.037138e-09
#>
#> Normed MSE value of iteration 8 is:
#> [1] 1.02442e-09
#>
#> Normed MSE value of iteration 9 is:
#> [1] 1.004947e-09
#>
#> Normed MSE value of iteration 10 is:
#> [1] 1.002143e-09
#> Skipping saving of results.
## Deconvolute only the first spectrum of the folder "bruker/sim_subset" into
evalwith(answers = expected_answers[-(3:4)], {
sim_decon <- MetaboDecon1D(sim, filename = "sim_01")
})
#> Start deconvolution of sim_01:
#> What is the name of the subfolder of your filepath:
#> [e.g. 10 for C:/Users/Username/Desktop/spectra_folder/spectrum_name/10]
#> 10
#> What is the name of the subsubsubfolder of your filepath:
#> [e.g. 10 for C:/Users/Username/Desktop/spectra_folder/spectrum_name/10/pdata/10]
#> 10
#> Water artefact fully inside red vertical lines? (y/n):
#> y
#>
#> Normed MSE value of iteration 1 is:
#> [1] 4.996559e-09
#>
#> Normed MSE value of iteration 2 is:
#> [1] 2.17297e-09
#>
#> Normed MSE value of iteration 3 is:
#> [1] 1.778563e-09
#>
#> Normed MSE value of iteration 4 is:
#> [1] 1.652956e-09
#>
#> Normed MSE value of iteration 5 is:
#> [1] 1.571827e-09
#>
#> Normed MSE value of iteration 6 is:
#> [1] 1.539285e-09
#>
#> Normed MSE value of iteration 7 is:
#> [1] 1.507361e-09
#>
#> Normed MSE value of iteration 8 is:
#> [1] 1.491203e-09
#>
#> Normed MSE value of iteration 9 is:
#> [1] 1.476108e-09
#>
#> Normed MSE value of iteration 10 is:
#> [1] 1.460549e-09
#> Save results as text documents at /home/runner/work/_temp/Library/metabodecon/example_datasets/bruker/sim_subset? (y/n)
#> n
#> Skipping saving of results.
#> Start deconvolution of sim_02:
#>
#> Normed MSE value of iteration 1 is:
#> [1] 4.449633e-09
#>
#> Normed MSE value of iteration 2 is:
#> [1] 1.688869e-09
#>
#> Normed MSE value of iteration 3 is:
#> [1] 1.276977e-09
#>
#> Normed MSE value of iteration 4 is:
#> [1] 1.135132e-09
#>
#> Normed MSE value of iteration 5 is:
#> [1] 1.085964e-09
#>
#> Normed MSE value of iteration 6 is:
#> [1] 1.056171e-09
#>
#> Normed MSE value of iteration 7 is:
#> [1] 1.037138e-09
#>
#> Normed MSE value of iteration 8 is:
#> [1] 1.02442e-09
#>
#> Normed MSE value of iteration 9 is:
#> [1] 1.004947e-09
#>
#> Normed MSE value of iteration 10 is:
#> [1] 1.002143e-09
#> Skipping saving of results.
## Deconvolute only the first spectrum of the folder "bruker/sim_subset" into
evalwith(answers = expected_answers[-(3:4)], {
sim_decon <- MetaboDecon1D(sim, filename = "sim_01")
})
#> Start deconvolution of sim_01:
#> What is the name of the subfolder of your filepath:
#> [e.g. 10 for C:/Users/Username/Desktop/spectra_folder/spectrum_name/10]
#> 10
#> What is the name of the subsubsubfolder of your filepath:
#> [e.g. 10 for C:/Users/Username/Desktop/spectra_folder/spectrum_name/10/pdata/10]
#> 10
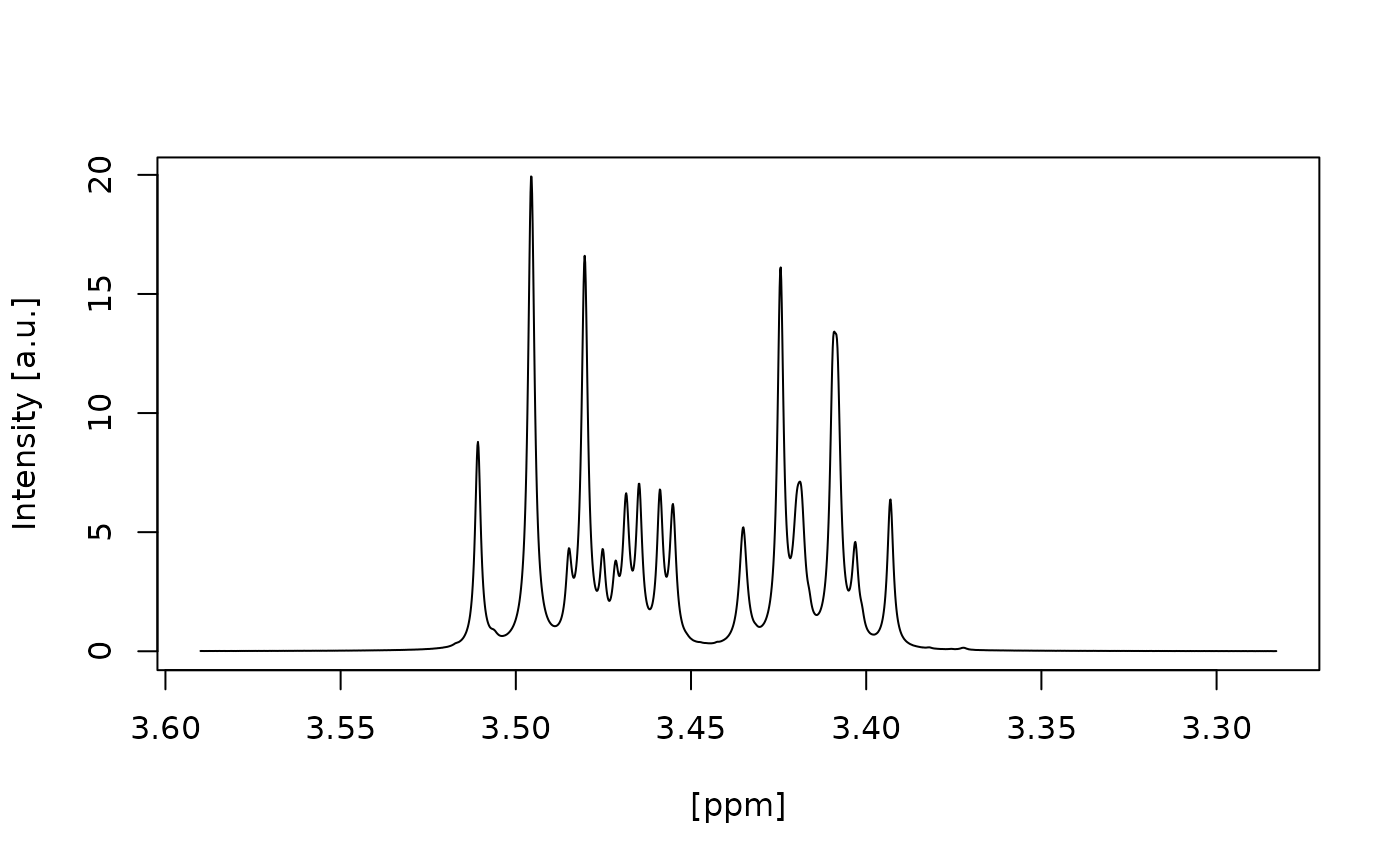 #> Signal free region borders correct selected? (Area left and right of the green lines) (y/n):
#> n
#> Choose another left border: [e.g. 12]
#> 3.55
#> Choose another right border: [e.g. -2]
#> 3.35
#> Signal free region borders correct selected? (Area left and right of the green lines) (y/n):
#> n
#> Choose another left border: [e.g. 12]
#> 3.55
#> Choose another right border: [e.g. -2]
#> 3.35
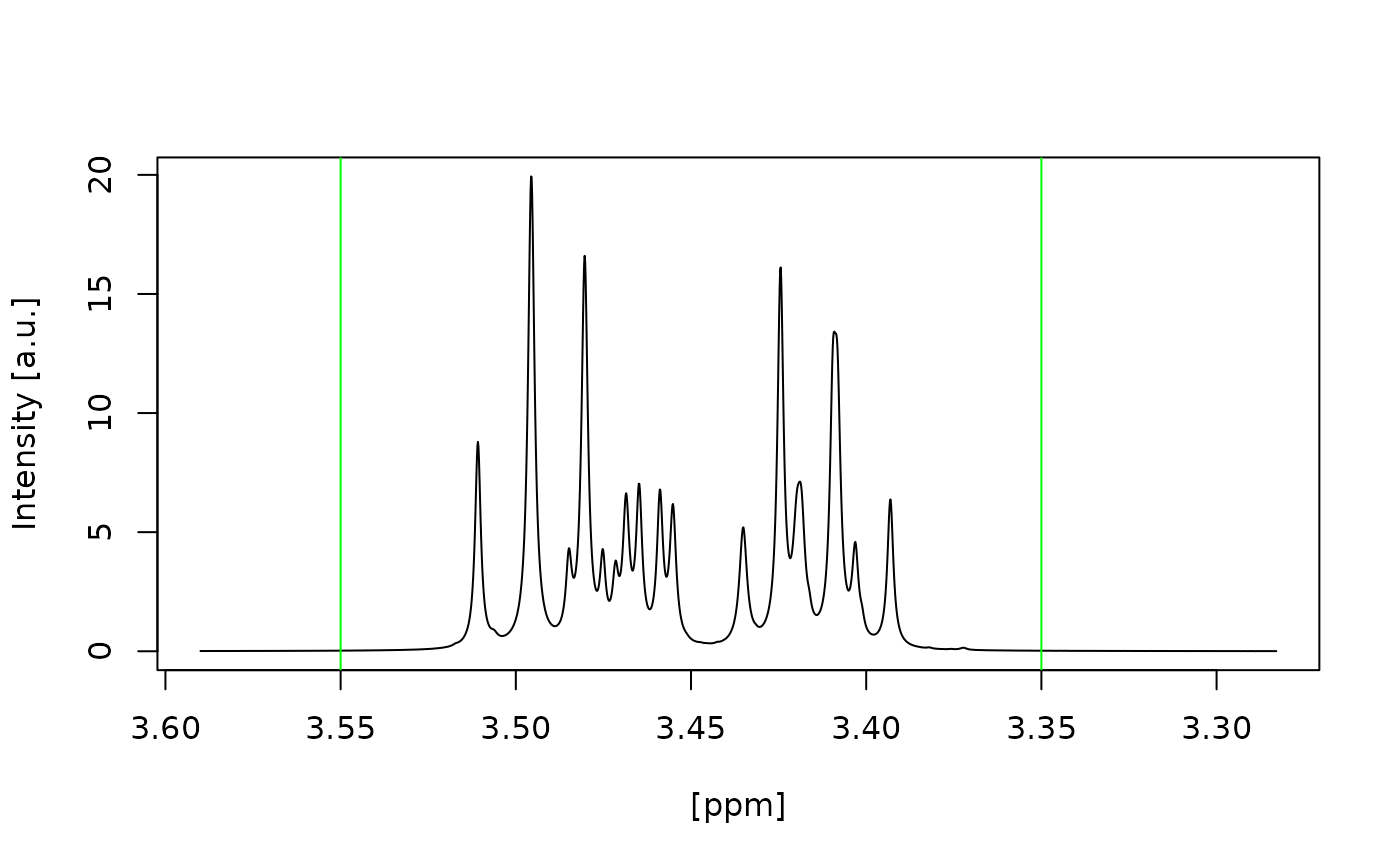 #> Signal free region borders correct selected? (Area left and right of the green lines) (y/n):
#> y
#> Signal free region borders correct selected? (Area left and right of the green lines) (y/n):
#> y
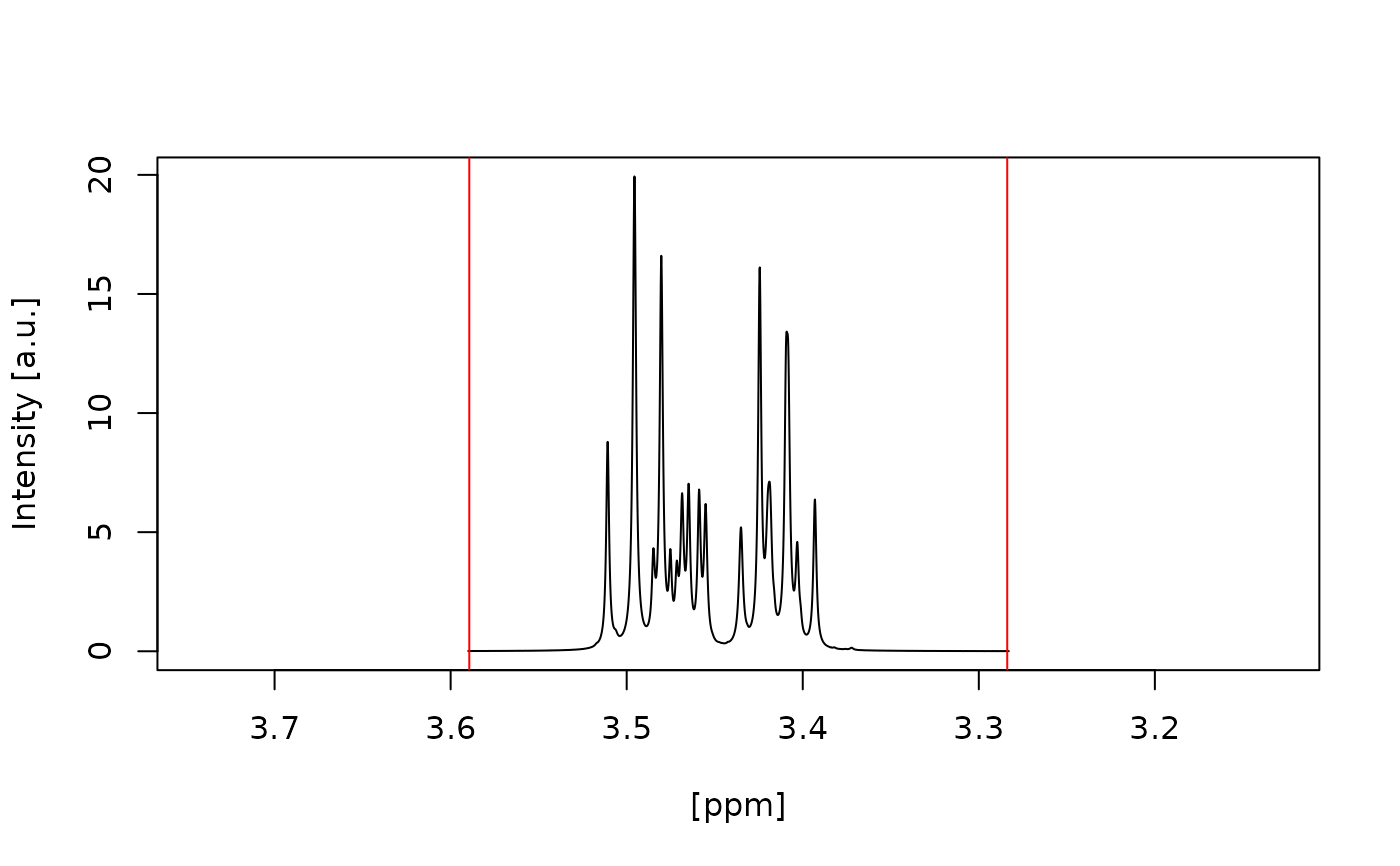 #> Water artefact fully inside red vertical lines? (y/n):
#> n
#> Choose another half width range (in ppm) for the water artefact: [e.g. 0.1222154]
#> 0
#> Water artefact fully inside red vertical lines? (y/n):
#> n
#> Choose another half width range (in ppm) for the water artefact: [e.g. 0.1222154]
#> 0
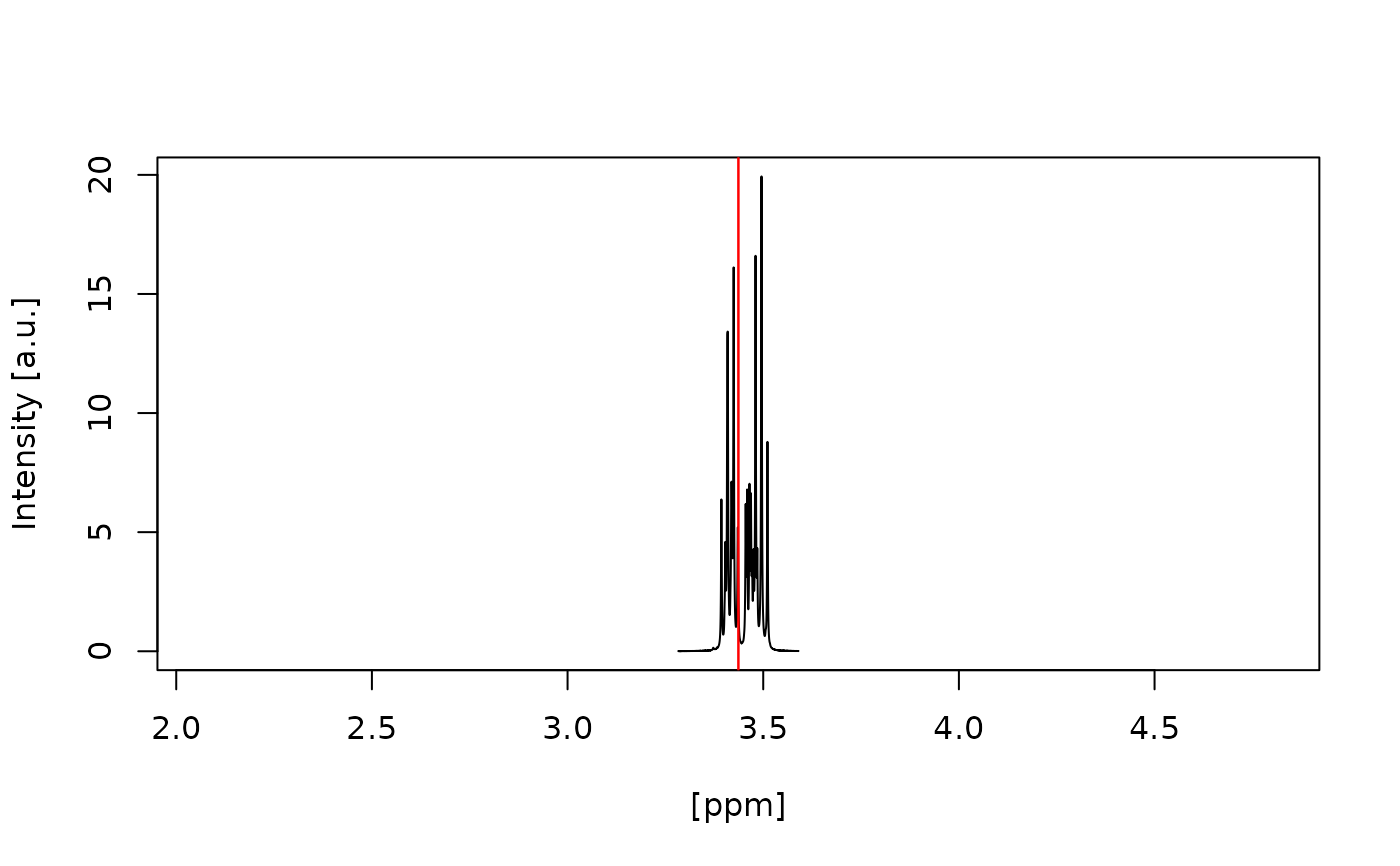 #> Water artefact fully inside red vertical lines? (y/n):
#> y
#>
#> Normed MSE value of iteration 1 is:
#> [1] 4.996559e-09
#>
#> Normed MSE value of iteration 2 is:
#> [1] 2.17297e-09
#>
#> Normed MSE value of iteration 3 is:
#> [1] 1.778563e-09
#>
#> Normed MSE value of iteration 4 is:
#> [1] 1.652956e-09
#>
#> Normed MSE value of iteration 5 is:
#> [1] 1.571827e-09
#>
#> Normed MSE value of iteration 6 is:
#> [1] 1.539285e-09
#>
#> Normed MSE value of iteration 7 is:
#> [1] 1.507361e-09
#>
#> Normed MSE value of iteration 8 is:
#> [1] 1.491203e-09
#>
#> Normed MSE value of iteration 9 is:
#> [1] 1.476108e-09
#>
#> Normed MSE value of iteration 10 is:
#> [1] 1.460549e-09
#> Save results as text documents at /home/runner/work/_temp/Library/metabodecon/example_datasets/bruker/sim_subset? (y/n)
#> n
#> Skipping saving of results.
#> Water artefact fully inside red vertical lines? (y/n):
#> y
#>
#> Normed MSE value of iteration 1 is:
#> [1] 4.996559e-09
#>
#> Normed MSE value of iteration 2 is:
#> [1] 2.17297e-09
#>
#> Normed MSE value of iteration 3 is:
#> [1] 1.778563e-09
#>
#> Normed MSE value of iteration 4 is:
#> [1] 1.652956e-09
#>
#> Normed MSE value of iteration 5 is:
#> [1] 1.571827e-09
#>
#> Normed MSE value of iteration 6 is:
#> [1] 1.539285e-09
#>
#> Normed MSE value of iteration 7 is:
#> [1] 1.507361e-09
#>
#> Normed MSE value of iteration 8 is:
#> [1] 1.491203e-09
#>
#> Normed MSE value of iteration 9 is:
#> [1] 1.476108e-09
#>
#> Normed MSE value of iteration 10 is:
#> [1] 1.460549e-09
#> Save results as text documents at /home/runner/work/_temp/Library/metabodecon/example_datasets/bruker/sim_subset? (y/n)
#> n
#> Skipping saving of results.